- 인슐린 신호 전달
- 공복 상태에서 간은 포도당을 혈액으로 분비하여 혈당을 유지하고 포도당을 소비하는 조직에 연료를 공급합니다. 이 과정을 간 포도당 생산(HGP)이라고 하며, 지방 조직에서 유래한 지방산과 글리세롤을 사용하여 간 글리코겐의 분해(글리코겐 분해) 및 포도당의 새로운 합성(포도당 신생)을 포함합니다[3]. 음식 섭취 후 췌장 β 세포에서 분비되는 인슐린은 동화 작용을 촉진하고 이화 작용을 억제합니다. 포도당 대사 과정에서 인슐린은 골격근 및 지방 조직과 같은 포도당을 소비하는 여러 조직을 자극하여 포도당을 흡수한 다음 간, 골격근 및 지방 조직에서 글리코겐과 지질의 합성을 촉진합니다[4]. 또한 인슐린은 지방 조직에서 포도당 신생 유전자 및 지방 분해의 발현을 억제하여 HGP를 억제합니다[5]. 인슐린은 또한 췌장 α 세포에서 글루카곤 분비를 억제하고[6,7] 중추 신경계를 통해 식욕을 감소시킵니다. 본 연구에서는 골격근, 간, 지방 조직의 포도당 대사에서 인슐린의 역할에 초점을 맞춘다.
- 인슐린의 세포 내 기능은 인슐린 수용체 티로신 키나아제(IRTK)를 통해 매개됩니다(그림 1). 인슐린이 IRTK의 세포외 도메인에 결합할 때, 인슐린은 IRTK 티로신 잔기의 자가인산화를 초래하는 구조적 변화를 유도하고 인슐린 수용체 기질(IRS), 성장 인자 수용체 결합 단백질-2(GRB-2), GRB-10, SHC-형질전환 단백질(SHC) 및 SH2B 어댑터 단백질-2(SH2B-2)와 같은 포스포티로신 결합 단백질의 후속 활성화를 유도합니다[8]. 포도당 및 지질 대사에 대한 인슐린의 효과는 주로 IRS의 IRTK 유도 인산화에 의해 매개되며, IRS는 포스파티딜이노시톨-3-OH 키나아제(PI3K)를 모집하고 포스파티딜이노시톨-4,5-비스포스페이트(PIP2)에서 포스파티딜이노시톨-3,4,5-트리스포스페이트(PIP3)의 생산을 촉매합니다. PIP3에 의해 원형질막에 모집된 후, Akt는 3-phosphoinositide-dependent kinase-1 (PDK1) 및 rapamycin 복합체 2 (mTORC2)의 기계론적 표적에 의해 활성화(인산화)되고 골격근, 간 및 지방 조직을 포함한 대사 조직의 다양한 다운스트림 기질을 인산화하여 이러한 조직에서 인슐린 유도 영양소 예약을 유도합니다.
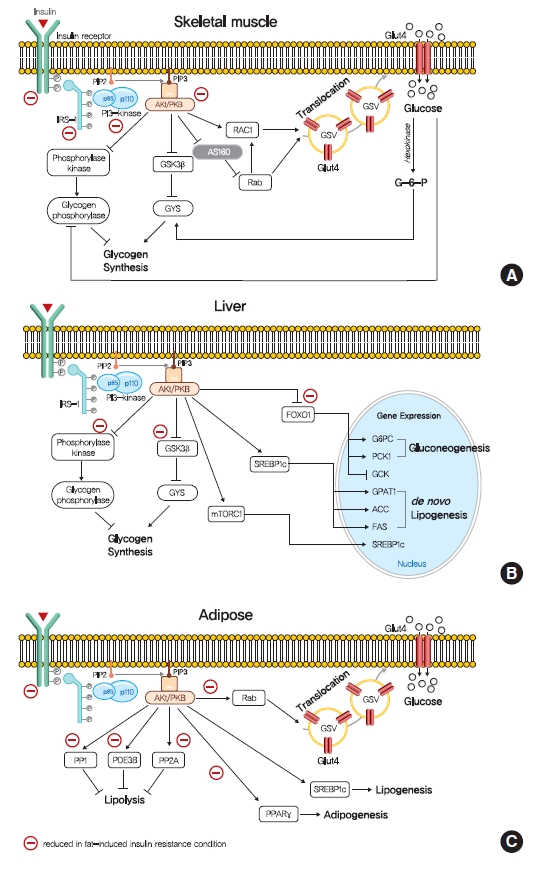
- 간, 골격근 및 지방 조직에서 인슐린 신호 전달의 역할. (A) 인슐린은 인슐린 수용체 티로신 키나아제(IRTK)에 결합하고 인슐린 수용체 기질-1(IRS-1)을 활성화하여 포스파티딜이노시톨-3-OH 키나아제(PI3K)를 모집하고 Akt를 활성화합니다. 골격근에서 Akt는 160kDa(AS160)의 GTPase 활성화 단백질(GAP) AKT 기질의 비활성화 및 Ras 관련 C3 보툴리눔 독소 기질 1(RAC1)의 GTP 결합 형태의 촉진에 의해 매개되는 원형질막으로의 포도당 수송체 4형(GLUT4) 저장 소포(GSV)의 전위를 통해 포도당 흡수를 촉진합니다. 인슐린은 글리코겐 합성효소 키나아제 3(GSK3) 억제 매개 글리코겐 합성효소(GYS) 활성화 및 포스포릴라아제 키나아제의 탈인산화를 통한 글리코겐 인산화효소 불활성화를 통해 글리코겐 합성을 자극합니다. (B) 간에서, Akt는 포크헤드 박스 O1 (FOXO1) 매개 글루코네오겐성 유전자 발현을 억제함으로써 글루코네오제네시스를 감소시킨다. 또한 인슐린은 GSK3와 단백질 인산가수분해효소 1(PP1)을 통해 GYS2와 글리코겐 인산화효소를 조절하여 간 글리코겐 합성을 증가시킵니다. 또한 인슐린은 스테롤 조절 요소 결합 단백질 1c(SREBP-1c)를 상향 조절하여 간을 신생 지방 형성을 증가시킵니다. (C) 백색 지방세포에서 인슐린은 지방 분해를 억제하여 포스포디에스테라아제 3B(PDE3B), PP1 및 단백질 인산가수분해효소-2A(PP2A)에 의해 매개되는 것으로 여겨지는 글루코네오겐 기질을 감소시켜 간 포도당 생성을 억제합니다. 인슐린은 또한 포도당 수송, 지방 형성 및 지방 형성을 촉진합니다. PIP2, 포스파티딜이노시톨-4,5-비스포스페이트; PIP3, 포스파티딜이노시톨-3,4,5-트리스포스페이트; PKB, 단백질 키나아제 B; mTORC1, 라파마이신 복합체 1의 기계론적 표적; G6PC, 포도당 -6- 포스파타아제; PCK1, 포스포에놀피루베이트 카르복시키나제 1 (PEPCK); GCK, 글루코키나아제; GPAT1, 글리세롤-3-포스페이트 아실트랜스퍼라제 1; ACC, 아세틸-CoA 카르복실라아제; FAS, 지방산 합성 효소.
- 골격근에서 인슐린 신호전달은 포도당 흡수와 순 글리코겐 합성을 촉진합니다(그림 1A). 인슐린은 골격근의 원형질막에 대한 포도당 수송체 4형(GLUT4) 저장 소포(GSV)의 고도로 조정된 전위 및 융합을 통해 포도당 수송 활성을 증가시킵니다[10]. 인슐린 신호전달에 의해 활성화된 후 Akt는 AS160(160kDa의 GTPase 활성화 단백질[GAP] AKT 기질, TBC1D4라고도 함)을 비활성화하여 소포 이동을 제어하는 작은 Rab GTPase 단백질 스위치를 활성화합니다[10]. 인슐린 유도 Akt는 또한 피질 액틴 재편성을 유도하여 GLUT4 전위를 촉진하는 Ras 관련 C3 보툴리눔 독소 기질 1(RAC1)의 구아노신 삼인산(GTP) 결합 형태를 촉진합니다[11]. 한편, 인슐린은 또한 글리코겐 분해를 억제하고 글리코겐 합성을 촉진함으로써 골격근의 순 글리코겐 합성을 조절합니다. 인슐린 신호전달은 Akt[12]에 의한 글리코겐 합성효소 키나아제 3(GSK3)의 인산화를 통해 글리코겐 합성효소(GYS)의 활성을 촉진하고 단백질 인산가수분해효소 1(PP1)의 활성화를 촉진하여 GYS의 탈인산화를 촉진합니다[13]. 또한 인슐린은 인산화효소 키나아제의 탈인산화를 통해 글리코겐 인산화효소 활성을 조절합니다[14].
- 간의 인슐린은 IRS1과 IRS2를 인산화하는 IRTK를 활성화하고, 궁극적으로 Akt2를 활성화하여 HGP를 감소시키고, 글리코겐 합성을 촉진하며, 전사적으로 지방 형성을 활성화합니다(그림 1B). 간 인슐린 신호전달의 주요 기능은 핵에서 FOXO1을 배제하는 포크헤드 박스 O1(FOXO1)[15]의 Akt 유도 인산화에 의해 매개되는 포도당 신생생성을 억제하여 HGP를 감소시키는 것이며, 따라서 포도당-6-인산가수분해효소(G6PC) 및 포스포에놀피루베이트 카르복실라제(PEPCK)와 같은 포도당 신생 유전자 발현의 전사 활성화를 방지합니다[15,16]. 인슐린은 포도당 신생 유전자 발현을 억제하는 것 외에도 지방 세포 지방 분해를 억제하여 간 포도당 신생 기질 수준을 감소시킵니다[17]. 또한, 인슐린은 HGP를 억제하는 것 외에도 골격근에서 발생하는 것처럼 GSK3 및 PP1을 통해 GYS(특히 간의 GYS2)와 글리코겐 인산화효소를 조절하여 간 글리코겐 합성을 증가시킵니다[18]. 또한 인슐린은 스테롤 조절 요소 결합 단백질 1c(SREBP-1c, 간 de novo 지방 형성의 마스터 전사 조절자)를 상향 조절하여 지질 동화 작용을 활성화한 후 아세틸-CoA 카르복실라제 1(ACC1), 지방산 합성효소(FAS) 및 글리세롤-3-포스페이트 아실전이효소 1(GPAT1)을 포함한 여러 지질 생성 유전자의 전사를 향상시킵니다[19,20].
- 백색 지방세포 조직에서 인슐린의 가장 중요한 생리적 기능은 지방 분해를 억제하는 것이며, 이는 다시 글루코네오겐성 기질을 감소시켜 HGP를 억제합니다(그림 1C)[21]. 인슐린 유도 지방 분해 억제에 책임이 있는 메커니즘은 완전히 이해되지 않았지만 고리형 아데노신 모노포스페이트(cAMP) 의존성 단백질 키나아제 A(PKA) 활성의 감소를 통해 포스포디에스테라아제 3B(PDE3B)에 의해 매개되는 것으로 믿어집니다[22]. 또한, PP1 및 단백질 인산가수분해효소-2A(PP2A)는 지분해 조절 단백질의 탈인산화를 통해 PI3K 의존성 인슐린 유도 지분해 억제를 매개하는 것으로 보입니다[23,24]. 인슐린은 소포 테더링, 도킹 및 융합과 관련된 표적의 인산화를 신호화하여 포도당 수송을 촉진하지만 전신 포도당 처리에 대한 기여도는 비교적 적습니다[25]. 인슐린은 또한 SREBP-1c를 활성화하고, 포도당 또는 지방산 수송 단백질(FATP)의 전좌를 신호하고, 지방산 에스테르화를 촉진하고[26], 전사 인자인 과산화소체 증식제 활성화 수용체 γ(PPARγ)를 통해 지방 형성을 자극하여 백색 지방 조직의 지방 형성을 촉진합니다[27].
- 인슐린 저항성
- 위에서 언급했듯이 인슐린 저항성은 생리학적으로 일부 유형의 조직이 정상적인 인슐린 수치에 반응하지 못하는 것으로 정의되므로 인슐린의 정상적인 기능을 유지하기 위해서는 정상 수치보다 높은 인슐린 수치가 필요합니다. 특히, HGP 및 지방 분해의 억제, 혈장 포도당의 세포 흡수 및 순 글리코겐 합성과 같은 인슐린의 포도당 조절 효과는 정상적인 혈장 수준의 인슐린 저항성 조직에서 관찰되지 않습니다[5]. 골격근은 인슐린 자극 포도당 처리를 위한 정량적으로 중심 조직이고, 간과 지방 조직은 포도당 유도 인슐린 신호 전달에 질적으로 중요한 부위이기 때문에 이러한 조직은 인슐린 저항성을 담당하는 메커니즘을 이해하는 데 중요한 것으로 간주됩니다.
- 인슐린 자극 포도당 소비는 주로 골격근에서 이루어지므로 근육 인슐린 저항성은 전신 대사에 영향을 미칠 수 있습니다[28]. IRTK 또는 GLUT4의 근육 특이적 결실에 의한 근육 특이적 인슐린 저항성은 간성 지방증에 대한 감수성을 증가시키고 동물 모델에서 비만을 증가시켰다[29,30]. 많은 분자 연구에서 인슐린 자극 근육 포도당 흡수는 GLUT4 전좌 장애로 인한 인슐린 저항성에 매우 취약하다는 것을 입증했습니다. 그러나 GLUT4의 원형질막으로의 전위와 T2DM에서의 포도당 수송은 AMP 활성화 단백질 키나아제(AMPK)에 의한 GSV 전좌 조절을 통한 저산소증 또는 운동에 의해 자극되며[31,32], 이는 인슐린 저항성에서 포도당 수송의 결함은 수송 시스템 자체의 결함이라기보다는 인슐린 신호 전달 경로의 이상임을 시사합니다. 또한, 골격근에서의 인슐린 저항성은 인슐린 신호전달의 근위 수준에서의 결함, 예를 들어, IRTK, IRS1, PI3K, 및 AKT의 활성에 의해 야기될 수 있다(도 1A). 또한 비만 마우스와 비만/당뇨병 환자에서 IRTK의 티로신 키나아제 활성이 골격근에서 감소하여 이 제안을 뒷받침합니다[33]. 또한 IRS1 티로신 인산화 및 IRS1 관련 PI3K 활성은 인슐린 저항성 골격근에서 감소하는 것으로 밝혀졌습니다[34].
- 간은 HGP를 억제하고 포도당을 글리코겐으로 침착시키는 것을 자극하여 식후 탄수화물 수치를 매우 중요하게 조절하며, 단식 중 포도당 생성의 주요 공급원입니다[35]. T2DM 환자에서 인슐린은 간 글리코겐 합성 또는 포도당 생성을 조절할 수 없으며, 간 포도당 신생 증가는 T2DM에서 공복 고혈당증의 주요 원인입니다[36,37]. 인슐린 저항성에서 간 포도당 신생생성의 결함 억제는 주로 지방 조직의 지방 분해 결함 및 간에서 FOXO1 전사 인자의 억제 해제와 관련이 있습니다[35]. 한편, 인슐린 저항성은 인슐린에 의한 글리코겐 합성 자극의 결함과도 관련이 있는데, 예를 들어 T2DM 환자는 공복 및 식후 간 글리코겐 함량이 낮다[38]. 또한, 간 인슐린 저항성은 공복과 수유에 의해 유도되는 간 글리코겐 대사의 진폭을 감소시킨다[36].
- 흥미롭게도, 모든 인슐린 기능이 인슐린 저항성이 있을 때 덜 반응하는 것은 아닙니다. 고인슐린혈증의 일부 인슐린 경로는 인슐린에 매우 민감하게 반응하는데, 이러한 현상을 선택적 인슐린 저항성 또는 경로 선택적 인슐린 반응성이라고 합니다[39]. 간 인슐린 저항성의 경우 인슐린은 HGP를 억제하지 못하고 지방 형성을 자극하여 고혈당증, 고지혈증 및 간 지방증을 초래합니다[39]. 선택적 인슐린 저항성의 근본적인 메커니즘은 확립되지 않았지만 몇 가지 가설이 제시되었습니다. 하나는 포도당 신생과 지방 형성 사이의 AKT 인산화의 기질 특이성의 포도당 처리 전위 차이와 관련이 있습니다[40]. Akt Ser473 인산화는 FOXO와 같은 포도당 신생과 관련된 일부 AKT 기질을 활성화할 수 있으며, 이러한 활성화는 인슐린 저항성의 배경에서 억제될 수 있는 반면, Thr308에서 AKT 인산화를 필요로 하는 GSK3β 및 결절성 경화증 복합체 2(TSC2)와 같은 다른 Akt 기질은 방해받지 않을 수 있습니다[41,42]. 간에서 선택적 인슐린 저항성의 또 다른 가능한 메커니즘은 인슐린 유도 SERBP-1c 활성화 및 포도당 신생 억제의 다양한 고유 민감도와 관련이 있으며, 이는 이러한 기능이 특정 인슐린 수준을 필요로 함을 시사합니다. 최근 연구에 따르면 인슐린 조절 간 지방 형성은 gluconeogenesis가 아닌 pleckstrin homology domain leucine-rich repeat protein phosphatase-2 (PHLPP-2)를 안정화하여 인슐린 자극 Akt 활성을 종료합니다 [43,44]. 이 보고서는 고지방 식이요법(HFD)이 PHLPP-2 안정성을 감소시켜 Akt 탈인산화를 지연시킨 다음 인슐린 펄스 후 말기에 장기간의 Akt 활성화를 유도한다는 것을 보여줍니다[43,44]. 식후 초기 단계의 Akt 활성의 증가는 HGP를 감소시키는 데 필요하고 후기 단계의 Akt 활성은 새로운 지방 형성을 증가시키기에 충분하지만 HGP는 증가시키지 않기 때문에[40,43,44], 종결되지 않은 장기간의 Akt 활성은 새로운 지방 형성을 향상시킬 수 있지만 HGP를 억제하지는 않습니다. 다른 가설은 간에서 선택적 인슐린 저항성이 인슐린 독립적 지방 형성에 의해 발생한다고 가정합니다. 영양소에 의한 지방 형성 유도는 탄수화물 반응 요소 결합 단백질(ChREBP), PPARγ coactivator 1-β 및 간 X 수용체 매개 SREBP-1c에 의해 매개되며[45-47], 이러한 대체 지방 형성 경로는 과당 및 단당류에 의해 활성화되는 것으로 나타났습니다[48,49].
인슐린 신호전달과 인슐린 저항성

골격근의 인슐린 저항성
간과 지방 조직의 인슐린 저항성
선택적 인슐린 저항성
- 많은 역학 연구에서 비만이 T2DM의 주요 위험 요인이라는 결론을 내렸으며, 비만인 사람은 T2DM에 걸릴 가능성이 최대 80배 더 높다고 합니다[50]. 또한, 연령 및 체질량 지수 일치 대조군에서 혈장 지방산 농도와 인슐린 감수성 사이에는 반비례 상관관계가 존재하는 것으로 나타났습니다[51]. 이러한 결과와 다른 결과들은 비만으로 인한 과도한 지질 함량이 인슐린 저항성과 T2DM의 주요 원인임을 보여줍니다. 과도한 지질 가용성으로 인한 인슐린 저항성의 발달을 담당하는 메커니즘에 대해 많은 이론이 제안되었습니다.
- 포도당-지방산 회로(Randle 회로)
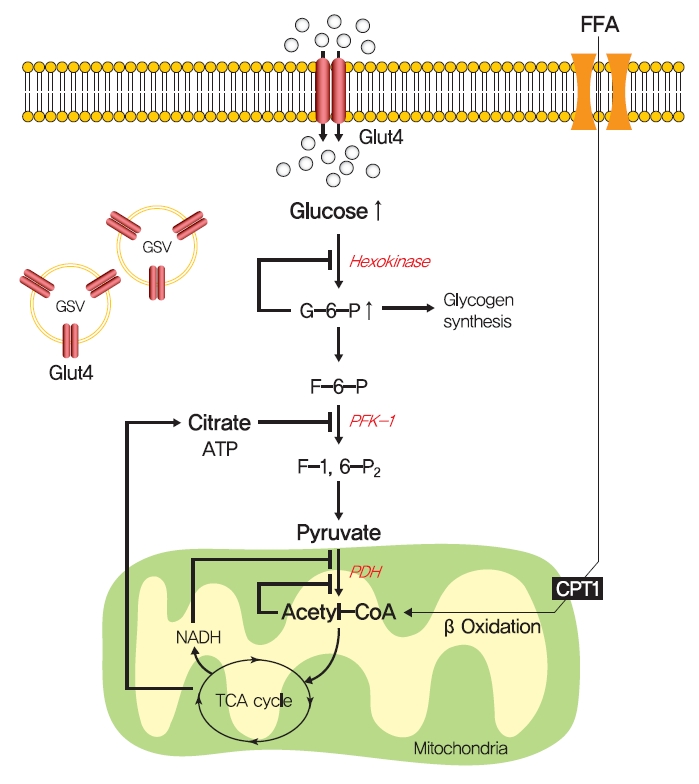
- 1963년, Randle 등[52]은 포도당 흡수의 결함을 특징으로 하는 골격근의 지질 유도 인슐린 저항성이 포도당-지방산 또는 Randle 회로라고 하는 지방산 산화 증가로 인한 제한된 인슐린 자극 포도당 이용에 기인한다는 가설을 세웠습니다. 비만에서 지방산 산화가 증가하면 주요 해당효소의 활성을 억제하여 포도당을 연료원으로 사용하는 것이 손상된다고 주장되었습니다. Randle et al. [52]에 따르면, 지방산 산화는 β산화를 통해 미토콘드리아 아세틸-CoA 수준을 증가시키고 이어서 피루브산 탈수소효소를 비활성화할 수 있으며, 이는 차례로 세포 내 구연산염 수준을 증가시키고 포스포프룩토키나제(주요 해당효소)를 억제하며, 헥소키나아제 활성을 억제하고 내세포 포도당의 축적 및 포도당 흡수 감소를 유발하는 트라미오세포 포도당-6-포스페이트의 축적을 유도할 수 있습니다(그림 2). 또한, 일부 실험 결과는 지방산의 주입이 쥐의 심장 및 횡격막 근육에서 근세포 포도당 이용을 감소시키고 근세포 내 포도당-6-인산염 농도를 증가시킨다는 것을 일관되게 보여주었습니다[53-55].
- 인슐린 저항성의 포도당-지방산 회로 가설. Randle 등[52]은 골격근의 지질 유도 인슐린 저항성이 지방산 산화 증가로 인한 제한된 인슐린 자극 포도당 이용에 기인한다고 제안했습니다. 지방산 산화는 미토콘드리아 아세틸-CoA 수치를 증가시키고 피루브산 탈수소효소(PDH)를 비활성화할 수 있으며, 이는 다시 세포 내 구연산염 수치를 증가시키고 포스포프룩토키나제 1(PFK-1)을 억제하며, 헥소키나아제 활성을 억제하고 세포내 포도당의 축적 및 포도당 흡수 감소를 유발하는 intramycellular glucose-6-phosphate(G6-P)의 축적을 유도합니다. FFA, 유리 지방산; GLUT4, 포도당 수송체 유형 4; GSV, GLUT4 저장 소포; ATP, 아데노신 삼인산; CPT1, 카르니틴 팔미토일전이효소 1; NADH, 감소된 니코틴아미드 아데닌 디뉴클레오티드; TCA, 트리클로로 아세트산. Figure 다운로드
- 포도당-지방산 회로는 포도당-6-인산염 및 글리코겐 합성 수준이 지방산에 의해 증가되어야 하고 해당과정이 억제되어야 함을 보여줍니다. 그러나 높은 혈장 지방산 농도에 노출되면 건강한 사람의 근육에서 인슐린 저항성이 발생하고 포도당-6-인산염 농도가 감소했습니다[56]. 또한, 만성 인슐린 저항성이 있는 T2DM 환자에서 인슐린 자극 근육 글리코겐 합성 및 포도당 산화가 억제되는 것으로 밝혀졌습니다[57,58]. 이러한 연구는 포도당-지방산 회로가 인슐린 저항성의 모든 장애를 완전히 설명할 수 없다는 것을 보여줍니다.
- 헥소사민 생합성 경로
- 포도당-지방산 회로가 지질 유도 인슐린 저항성을 만족스럽게 설명하지는 못하지만, 증거에 따르면 근육의 아세틸-CoA 및 구연산염 수치는 지방 산화 증가에 따라 증가하며, 이는 근육의 포도당-6-인산염 수치와 독립적으로 포도당 수송을 조절하는 또 다른 경로를 시사합니다(그림 3). 헥소사민 생합성 경로(HBP)는 인슐린 저항성 발생에 대한 또 다른 설명을 제공합니다. Fructose-6-phosphate는 주로 glucose-6-phosphate에서 생성되며 해당과정 동안 fructose-1,6-bisphosphate로 우선적으로 대사되지만, fructose-6-phosphate의 약 5%는 HBP의 속도 제한 효소인 glutamine:fructose-6-phosphate amidotransferase (GFAT)에 의해 글루코사민-6-phosphate로 전환됩니다[59]. 글루코사민-6-포스페이트는 지질과 단백질의 당화(glycosylation) 및 O-GlcNA실화(O-GlcNAcylation)를 위한 공여체 당 뉴클레오티드 역할을 하는 우리딘 5'-디포스페이트 N-아세틸글루코사민(UDP-GlcNAc)으로 전환되며[59], 이러한 변형, 특히 O-GlcNAcylation은 유전자 발현 또는 효소 활성을 조절하여 표적 단백질에 영향을 미칠 수 있습니다[60].
- 인슐린 저항성의 헥소사민 생합성 경로(HBP). HBP에서 과당-6-인산염(F-6-P)은 글루타민:과당-6-인산염 아미도전이효소(GFAT)에 의해 글루코사민-6-인산(글루코사민-6-P)으로 전환되고, 글루코사민-6-P는 지질과 단백질의 O-GlcNAcylation을 위한 공여당 뉴클레오티드 역할을 하는 우리딘 5'-디포스페이트 N-아세틸글루코사민(UDP-GlcNAc)으로 전환됩니다. O-GlcNAcylation은 유전자 발현 또는 효소 활성을 조절하여 표적 단백질에 영향을 미칠 수 있습니다. 인슐린 신호 전달 경로 구성 요소, 포유류 비조정-18c(Munc18-c) 및 포크헤드 박스 O1(FOXO1)은 O-GlcNAc로 변형됩니다. PIP2, 포스파티딜이노시톨-4,5-비스포스페이트; PIP3, 포스파티딜이노시톨-3,4,5-트리스포스페이트; IRS-1, 인슐린 수용체 기질-1; PKB, 단백질 키나아제 B; GSK3, 글리코겐 합성효소 키나아제 3; GSV, GLUT4 저장 소포; GLUT4, 포도당 수송체 유형 4; GYS, 글리코겐 합성 효소; OGT, O-GlcNAc 전이효소; OGA, O-GlcNA케이스; ATP, 아데노신 삼인산; PFK-1, 포스포프룩토키나제 1; PDH, 피루브산 탈수소효소; NADH, 감소된 니코틴아미드 아데닌 디뉴클레오티드; TCA, 트리클로로 아세트산. Figure 다운로드
- 30년 전, Marshall 등[61]은 O-GlcNAcylation이 인슐린 감수성을 조절한다고 처음으로 제안했으며, O-diazoacetyl-L-serine(아자세린) 또는 6-diazo-5-oxonorleucine(DON)과 같은 아미도전이효소 억제제를 사용하여 GFAT를 억제함으로써 포도당 유도 인슐린 저항성의 영향을 차단할 수 있음을 보여주었습니다. 그 후, 글루코사민이 인슐린 저항성을 유도한다는 것이 확인되었습니다[62,63]. 또한, 고혈당증 및 고인슐린혈증은 골격근에서 UDP-GlcNAc 수치를 증가시키는 것으로 보고되었으며[64,65], O-GlcNAc 수치는 당뇨병 또는 인슐린 저항성 쥐의 각막, 췌장 및 골격근에서 상승하는 것으로 보고되었습니다[66,67]. 또한, 지방 조직 또는 골격근에서 GFAT 과발현은 생쥐에서 포도당 처리의 감소를 특징으로 하는 전신 인슐린 저항성을 발달시켰습니다[68]. 최근 HFD에서 골격근 특이적 O-GlcNAc 전이효소(OGT) 녹아웃 마우스는 혈장 포도당 수치와 포도당 내성이 낮은 것으로 보고되었습니다[69]. 또한, 단백질에서 O-GlcNAc를 제거하는 O-GlcNAcase(OGA)의 과발현은 db/db 마우스에서 전신 포도당 내성 및 인슐린 감수성을 유의하게 개선시켰으며, O-(2-acetamido-2-deoxy-D-glucopyranosylidene) amino-N-phenylcarbamate(PUGNAc)(OGA 억제제)는 지방세포에서 인슐린 매개 포도당 흡수를 억제했습니다[70,71]. 또한, 인간 OGA 상동체인 수막종 발현 항원 5(MGEA5)의 단일 뉴클레오티드 다형성은 피마 인디언과 멕시코계 미국인의 T2DM 발병과 관련이 있었습니다[72,73].
- 인슐린 저항성에서 HBP의 분자 메커니즘은 완전히 확립되지 않은 반면, O-GlcNAcylation은 인슐린 저항성에서 HBP의 가장 수용 가능한 메커니즘을 제공합니다. 단백질의 O-GlcNAcylation은 단백질 활성과 신호 전달을 조절할 수 있는 부위에 대한 인산화와 경쟁하는 것으로 제안되었습니다[74]. 특히, IRS-1/2, PI3K, PDK1 및 Akt와 같은 인슐린 신호전달 경로 성분은 O-GlcNAc로 변형됩니다[75,76]. 인슐린 신호전달의 이러한 근위 성분 외에도 인슐린 자극 GLUT4 전좌에 필수적인 단백질인 포유류 비협조-18c(Munc18-c)는 글루코사민 유도 인슐린 저항성의 배경에서 O-GlcNAc가 변형된 것으로 보고되었으며, 간에서 글루코네오겐성 유전자의 마스터 전사 인자인 FOXO1도 당뇨병에서 O-GlcNacylated되었습니다[77,78].
- 인슐린 저항성에서 HBP의 역할에 대한 증거가 축적되고 있음에도 불구하고 논쟁은 계속되고 있습니다. Robinson 등[79]은 아데노바이러스에 의한 OGA 과발현 및 OGT 녹다운이 인슐린 자극에 의한 손상된 Akt 활성화를 회복시키고 3T3L1 지방세포에서 포도당 유도 인슐린 저항성을 예방하지 못한다는 것을 보여주었으며, 또 다른 연구에서는 OGA 억제제인 1,2-dideoxy-2'-propyl-α-ᴅ-glucopyranoso-[2,1, D]-Δ2'-thiazoline(NButGT)을 사용한 치료가 3T3-L1 지방세포에서 결함 있는 포도당 흡수 및 Akt 인산화를 특징으로 하는 인슐린 저항성을 유도하지 않았다[80]. 종합하면, in vivo 및 in vitro에서 HBP의 역할 간의 불일치는 HBP의 역할과 인슐린 저항성과 관련된 메커니즘을 결정하기 위해 in vivo 시스템을 사용하는 보다 철저하고 체계적인 접근이 필요하다는 것을 시사합니다.
- 이소성 지질 축적
- 비만은 T2DM의 명백하고 중요한 위험 요인이지만, 많은 연구에 따르면 말초 조직, 특히 간과 골격근에 이소성 지질이 축적되면 내장 지방이 없는 경우에도 더 심각한 인슐린 저항성을 유발할 수 있습니다[81]. 일관되게 T2DM이 있는 비만 환자의 70% 이상이 NAFLD를 가지고 있는 것으로 알려져 있으며, NAFLD 환자는 거의 보편적으로 T2DM과 간 인슐린 저항성을 가지고 있습니다[82]. 또한 적당한 체중 감량 또는 렙틴 치료를 통해 간내 트리글리세라이드 함량을 줄이는 중재는 T2DM, NAFLD 및 지방이영양증 환자에서 간 인슐린 저항성을 극적으로 역전시킵니다[83]. 또한, 간내 및 근세포내 지질 함량은 내장 지방 조직 부피보다 인슐린 저항성을 훨씬 더 잘 예측할 수 있는 것으로 간주되며, 이는 간과 골격근의 지질 축적 증가가 인슐린 신호 전달을 손상시키고 인슐린 저항성을 유발할 수 있음을 강력하게 시사합니다[84,85].
- 수많은 동물 실험에서 인슐린 저항성이 간이나 골격근의 이소성 지질 축적에 의해 발생한다는 개념을 뒷받침합니다. 여러 연구에 따르면 단기간의 HFD 공급 또는 지질/헤파린 주입으로 인한 간 및 골격근의 지질 축적은 쥐의 인슐린 저항성을 유발합니다[86]. 또한 간 또는 근육에서 지단백 리파아제(LPL)의 과발현은 말초 인슐린 저항성과 각 조직에 지질의 축적을 유도했으며[87,88], 골격근 특이적 LPL 결실은 HFD 도전 근육에서 인슐린 신호 전달을 강화했습니다[89]. 또한, CD36 또는 FATP-1과 같은 지방 수송 단백질을 삭제하면 골격근에서 인슐린 매개 포도당 흡수가 증가하고[90,91], FATP2 또는 FATP5의 간 특이적 녹다운은 HFD 유발 간동증을 현저히 감소시키고 포도당 내성을 증가시켰습니다[92,93].
- 지방이영양증은 고중성지방혈증, 말초이소성 지방 침착 및 심각한 인슐린 저항성을 유발하는 지방 조직의 지방 세포가 크게 감소하는 것이 특징이기 때문에 내장 지방이 인슐린 저항성에 기여하지 않는 경우 이소성 지질 축적의 역할을 밝히는 가장 잘 확립된 임상 증거를 제공합니다. 지방이영양증에서는 식후 지방산 유입이 지방 조직에 의해 완충되지 못하기 때문에 지방산이 간과 골격근을 포함한 다른 대사 조직으로 전달되어 인슐린 신호 전달을 손상시키고 심각한 인슐린 저항성을 유발합니다[94]. 유전적 지질영양성 마우스는 또한 간과 골격근의 이소성 지질 축적에 의해 유도된 인슐린 저항성을 나타낸다. 백색 지방 조직이 부족하여 A-ZIP/F-1 무지방 마우스라고 불리는 지방세포 특이적 염기성 부위-류신 지퍼(B-ZIP) 전사 인자 녹아웃 마우스는 근육 및 간에서 IRS-1 및 -2 관련 PI3K 활성의 심각한 결함으로 인해 고인슐린혈증 및 고혈당증입니다[95,96]. 또한, A-ZIP/F-1 무지방 마우스의 인슐린 기능은 깔짚 동료의 파라메트리컬 지방을 이식하여 극적으로 회복되었으며, 이는 골격근과 간으로의 트리글리세라이드 분포가 인슐린 저항성의 중요한 요인이 될 수 있음을 시사합니다[95]. 지방세포 분화를 억제하는 분비 단백질인 지방전세포 인자-1(Pref-1)의 과발현은 지방이영양증 모델의 특성, 즉 지방조직량 감소, 이상지질혈증, 인슐린 저항성을 유도했다[97,98]. 또한, de novo sphingolipid 합성의 첫 번째 단계를 촉매하는 serine palmitoyltransferase 2 (Sptlc2)의 지방 세포 특이적 녹아웃에 의한 de novo sphingolipid 생합성의 억제는 손상된 지방 조직 발달 및 전신 인슐린 저항성으로 진행된 지질영양성 표현형을 나타냈습니다[99]. 백색 지방세포에서 단안 지질 방울을 형성하는 능력은 백색 지방세포가 지질을 저장하는 능력을 유지하는 데 필요합니다. 지방 특이적 단백질 27(Fsp27)의 마우스를 녹아웃한 결과, 지방세포에서 다안 지질 방울이 나타나고 지방 분해가 증가하여 간 지방증과 인슐린 저항성이 나타났습니다[100]. 이 연구는 간과 근육에 과도한 지질 축적이 인슐린 저항성을 유발할 수 있음을 보여줍니다.
- 과도한 지질 축적이 간과 근육의 인슐린 신호 전달을 어떻게 방해하는지, 특히 전형적인 비만 관련 인슐린 저항성에서 표면 인슐린 수용체 하향 조절 및 손상된 인슐린 신호 전달과 관련하여 오랫동안 연구되어 왔습니다[101]. 이소성 지질 축적이 인슐린 저항성을 유발하는 메커니즘에 대한 가장 그럴듯한 가설은 디아실글리세롤(DAG), 리소포스파티드산(LPA), 세라마이드, 아실카르니틴을 포함한 여러 지질 대사 산물이 간과 골격근에서 인슐린 저항성의 발병에 관여한다는 것입니다.
- 지방산은 세포에서 지방 아실-CoA로 빠르게 에스테르화되고, 이는 글리세롤 골격으로 전달되어 지방 형성을 통해 LPA, DAG 및 트리아실글리세롤(TAG)을 형성합니다(그림 4). 이러한 지질 중간체, 특히 DAG는 인슐린 저항성의 발병기전과 관련된 주요 신호 전달 경로에서 두 번째 전달자로 기능할 수 있습니다[102]. 고지방 유발 및 유전적으로 비만인 설치류는 간 인슐린 저항성과 간 DAG 함량 상승을 나타냈으며[103,104], phorbol myristate acetate(PMA, DAG 유사체)로 처리한 간세포는 손상된 IRTK 티로신 키나아제 활성과 인슐린 자극 GYS 활성을 나타냈습니다[105].
- 인슐린 저항성에 대한 Diacylglycerol(DAG)-protein kinase C(PKC) 가설. 지방산은 세포에서 지방 아실-CoA로 빠르게 에스테르화되어 지방 형성을 통해 리소포스파티드산(LPA), DAG 및 트리아실글리세롤(TAG)을 형성합니다. 간 DAG 수치가 증가하면 nPKC(간 및 골격근의 PKCε 및 PKCθ)가 원형질막으로 전위되고 인슐린 수용체 티로신 키나아제(IRTK) 티로신 키나아제 활성이 Thr1160에서 인산화되어 인슐린 수용체 기질 2(IRS-2), 포스파티딜이노시톨-3-OH 키나아제(PI3K) 및 Akt2를 비활성화합니다. PIP2, 포스파티딜이노시톨-4,5-비스포스페이트; PIP3, 포스파티딜이노시톨-3,4,5-트리스포스페이트; PKB, 단백질 키나아제 B; PLC, 포스포리파제 C; FFA, 유리 지방산; MGL, 모노아실글리세롤 리파아제; MAG, 모노아실글리세롤; HSL, 호르몬 민감성 리파아제; ATGL, 지방 트리글리세라이드 리파아제; CGI-58, 비교 유전자 식별-58; GPAT, 글리세롤-3-포스페이트 아실트랜스퍼라제; AGPAT, 아실글리세롤포스페이트 아실트랜스퍼라제; PA, 포스파티드산; PAP, 포스파티드산 인산분해효소; DGAT, 디아실글리세롤 아실전이효소; ACS, 아실-CoA 합성효소; FAS, 지방산 합성 효소; ACC, 아세틸-CoA 카르복실라아제; CPT1, 카르니틴 팔미토일전이효소 1; TCA, 트리클로로 아세트산. Figure 다운로드
- 지질 유도 인슐린 저항성에 대한 DAG 가설은 활성화된 단백질 키나아제 C(PKC)에 의한 인슐린 신호 전달의 간섭이 인슐린에 민감한 조직 내에 DAG가 축적되어 발생한다는 것입니다(그림 4). PKC 계열에는 세 가지 그룹(전통적, 신규적, 비정형적)이 있으며, DAG에 대한 친화력이 훨씬 더 큰 새로운 PKC(nPKC)는 인슐린 저항성에서 DAG의 역할을 매개하는 것으로 알려져 있습니다[106,107]. 간에서 간 DAG 수치가 증가하면 PKCε(간의 1차 nPKC 동형)이 원형질막으로 전위되고 Thr1160에서 인산화되어 IRTK 티로신 키나아제 활성이 억제되어 IRS2, PI3K 및 Akt2의 인슐린 자극 인산화(활성화)가 감소했습니다[108,109]. 같은 맥락에서, 간 DAG 함량과 원형질막으로의 PKCε 전위는 인간의 세라마이드, 소포체(ER) 스트레스 마커 및 염증성 사이토카인 수치와 같은 다른 요인보다 간 인슐린 저항성의 더 강력한 예측 인자인 것으로 나타났습니다[106,110,111]. 간에서 관찰되는 것과 유사한 방식으로, intramycellular DAG의 축적은 Ser1101에서 IRS-1의 인산화를 유도하고 IRS-1의 인슐린 자극 인산화를 차단하는 PKCθ(muscle-type nPKC)[112,113]를 활성화하여 인슐린 신호 전달 및 근육 포도당 흡수를 손상시킵니다[114,115]. 대규모 연구는 또한 인슐린 저항성에 대한 DAG-PKC 유도 가설을 확증했습니다. 당뇨병 쥐의 가자미근의 막 분획과 비만 쥐의 간에서 높은 PKC 수치가 보고되었습니다[106,116]. 중요한 것은 HFD 섭식에 의해 유도된 고립된 간 지방증 및 간 인슐린 저항성이 있는 쥐에서 원형질막으로의 PKCε 전위가 관찰되었다는 것입니다[86,117]. 또한, 안티센스 올리고뉴클레오티드(ASO)를 사용한 PKCε의 녹다운은 지방 유발 간 인슐린 저항성으로부터 랫드를 보호했으며[109], PKCε 녹아웃 마우스(Prkce−/−) 마우스는 고지방 수유 7일 후 야생형 대조군보다 향상된 내당능을 보였습니다[118]. 아포지단백 CIII(Apolipoprotein CIII, ApoC3) 과발현은 식이 유발 간 지방증 및 간 인슐린 저항성을 촉진하여 DAG 및 막 국소화 PKCε를 높였습니다[119]. PKCε과 마찬가지로 PKCθ는 근아세포에서 인슐린 신호전달을 조절하고, PKC-θ knockout은 쥐 골격근에서 인슐린 신호전달 및 포도당 수송의 지방 유도 결함을 예방했습니다[120]. 또한, PKCθ 과발현은 C2C12 근아세포에서 인슐린 반응성 감소를 특징으로 하는 인슐린 저항성을 유발했습니다[121].
- 특히, 모든 DAG가 PKC 원형질막으로의 전위를 자극하고 인슐린 신호전달을 억제하여 T2DM을 유도할 수 있는 것은 아닙니다. DAG 및 DAG 입체이성질체 유형의 세포 내 국소화는 PKC를 활성화하고 인슐린 저항성을 유도하는 데 중요한 것으로 간주되었습니다. 3개의 DAG 입체이성질체 중에서, PIP2에 대한 포스포리파제 C의 작용의 산물인 sn-1,2-DAG는 PKC를 활성화하는 유일한 입체이성질체인 것으로 보고되었다[122]. 더욱이, 지질 방울 내에서의 DAG의 축적은 원형질막이 아니고, 인슐린 신호전달을 억제하지 않았으며, 지방 트리글리세라이드 리파아제 보조인자(comparative gene identification-58 [CGI-58]) ASO로 처리된 마우스에서 나타난 바와 같이, 간 인슐린 저항성을 유도하지 않았다. CGI-58 ASO를 처리한 마우스는 간 DAG 함량이 증가했지만 간 인슐린 감수성은 유지되었습니다[123]. 결과적으로, 원형질막의 sn-1,2-DAG 분포는 인슐린 신호전달을 방해하고 PKC 활성화를 통해 인슐린 저항성에 기여했습니다[123,124].
- 세라마이드는 세포 내 지방산과 스핑고신에서 생산되는 스핑고지질이자 필수 생체 활성 지질이며 지질 유도 인슐린 저항성을 매개하는 것으로 알려져 있습니다[125]. 세라마이드는 세포막 안정화에 중요한 역할을 하며 신호 분자의 분포를 조절합니다. 비만인에서 간 세라마이드는 인슐린 저항성(HOMA-IR) 점수에 대한 항상성 모델 평가와 관련이 있는 것으로 알려져 있으며[126], 인슐린 저항성이 있는 비만 쥐는 간 및 근육 세라마이드 함량이 지속적으로 증가하는 것으로 보고되었습니다[103]. 세린 팔미토일전이효소의 억제제인 미리오신(myriocin)을 사용한 세라마이드 합성 억제는 지방을 먹인 마우스에서 인슐린 저항성을 방지하고 세라마이드 함량을 약화시켰다[127,128]. 또한, 세라마이드 합성의 게이트키퍼인 디하이드로세라마이드 데사투라제 1(Des1)에 대한 이형접합 마우스는 공복 HOMA-IR 점수와 간 내 총 세라마이드 수치가 낮았습니다[127]. 또한 세라마이드 합성효소 6(CerS6)의 간 특이적 녹아웃은 간 세라마이드 수치(특히 C16:0 종)를 감소시키고, HFD로 인한 비만을 예방하며, 포도당 내성을 개선했습니다[129]. 또한 세라미다아제의 간 특이적 과발현은 HFD를 먹인 마우스를 간 지방증으로부터 보호하고, 간 인슐린 신호 전달을 강화하고, 16:0, 18:0 및 20:0 세라마이드를 포함한 일부 세라마이드 종의 수치를 감소시켰습니다[130]. 또한 지방 또는 간 특이적 아디포넥틴 수용체(AdipoR) 과발현은 총 세라마이드 함량을 감소시키고 HFD 유도 인슐린 저항성을 예방했습니다[131]. 이러한 연구의 결과는 세라마이드 수치가 지질 유도 인슐린 저항성에 영향을 미친다는 것을 시사합니다.
- 세라마이드가 인슐린 저항성을 유도하는 근본적인 분자 메커니즘은 명확하게 입증되지 않았습니다. 그러나, 일부 후보 기전, 즉 비정형 PKCζ의 활성화를 통한 AKT 전위의 손상과 PP2A의 활성화가 제안되었다[132,133]. 간 및 지방 조직에서 세라미다아제의 과발현은 PKCζ 활성을 감소시키고 세라마이드 유도 인슐린 저항성 및 간 지방증을 예방했습니다[130]. 그러나 세라마이드 유사체인 C2-세라마이드는 PP2A를 활성화했지만 PKCζ는 활성화하지 않았으며 갈색 지방세포에서 Akt 활성을 비활성화했으며[134], 이는 PP2A가 인슐린 저항성에서 세라마이드의 역할을 매개할 수 있음을 시사합니다. 그러나 LB1(PP2A 억제제)은 공복 혈장 포도당 상승으로 입증된 바와 같이 간 인슐린 저항성을 증가시켰고, PP2A 활성화가 인슐린 저항성의 주요 세라마이드 경로가 아님을 나타내는 간 인슐린 신호전달을 강화했음에도 불구하고 쥐의 고인슐린혈증-유혈당 클램프 테스트 중 포도당 주입 속도를 감소시켰습니다[135]]. 또한, 섬유아세포 성장인자 21(FGF21), 아디포넥틴 및 NOD 유사 수용체 피린 도메인 함유 단백질-3(NLRP-3) 인플라마좀이 세라마이드의 기능을 매개하거나 연관시키는 것으로 제안되었습니다[136-138]. 종합하면, 세라마이드가 인슐린 저항성을 유발할 수 있다는 증거가 늘어나고 있지만, 세라마이드가 인슐린 신호 전달 또는 포도당 수송 전위를 손상시키는 분자 메커니즘을 밝히기 위해서는 추가 연구가 필요합니다.
- 다른 메커니즘: ER 스트레스 및 염증
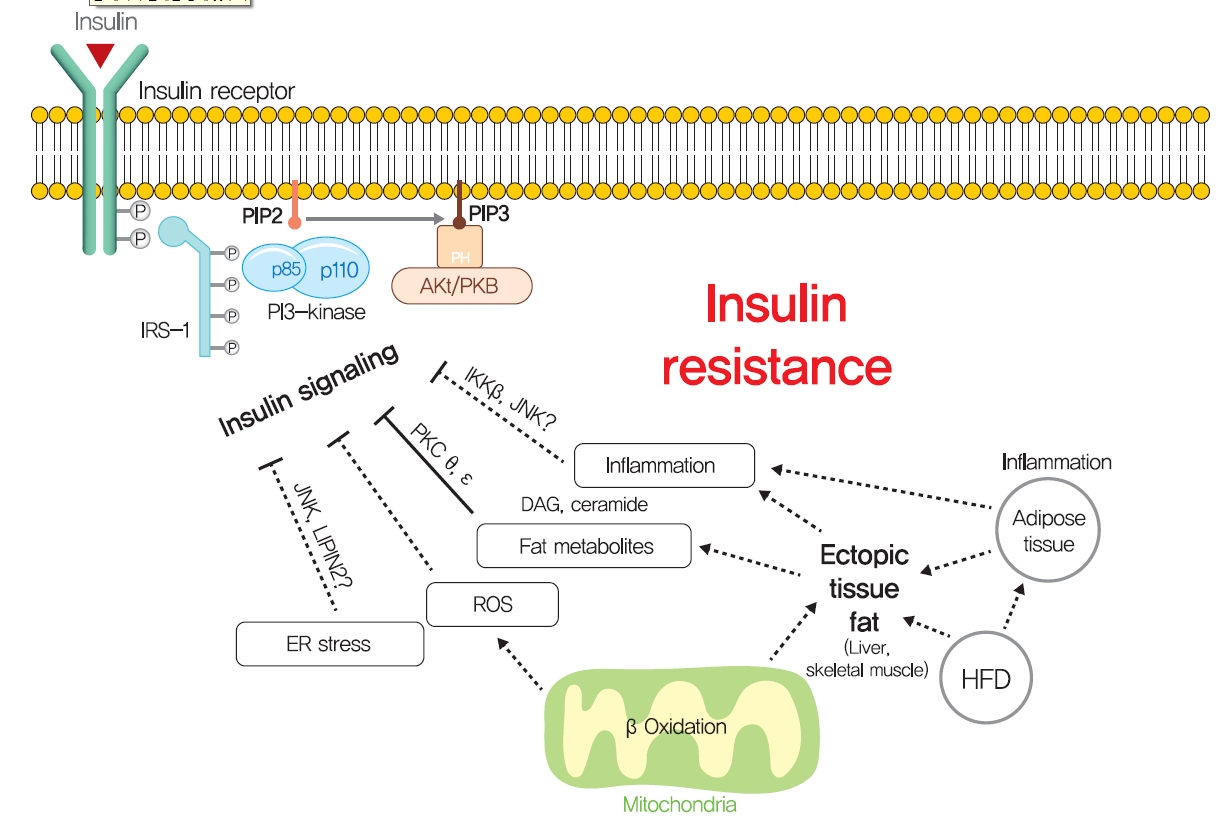
- 근육과 간의 이소성 지질 축적이 비만 또는 이상지질혈증 환자의 인슐린 저항성 발생을 그럴듯하게 설명하지만, 비만으로 인한 인슐린 저항성의 기전을 설명하기 위해 몇 가지 다른 가설도 제안되었습니다(그림 5). 특히, 응급실 스트레스는 간과 췌장 β 세포의 인슐린 저항성의 기초가 되는 것으로 제안되었습니다. 응급실 스트레스는 비만에 의해 강화되는 것으로 알려져 있다[139]. 영양 과잉 상태에 노출되면 간은 영양소를 처리하고 응급실에서 풀린 단백질을 축적하여 풀린 단백질 반응(UPR)을 향상시키기 위해 과도한 효소를 생산해야 합니다. 이 과정에서 포도당 조절 단백질 78(GPR78, BiP라고도 함)의 모집은 이노시톨 필요 효소-1(IRE1α), PKR 유사 ER 키나아제(PERK) 및 전사 인자 6(ATF6)의 활성화를 초래하여 단백질 번역을 억제하고 ER 샤페론을 촉진하여 풀린 단백질 수준을 감소시킵니다[140]. 실험적으로, 투니카마이신에 의해 유도된 ER 스트레스는 c-Jun N-말단 키나아제(JNK)에 의한 IRS-1의 세린 인산화를 통해 인슐린 신호 전달을 억제합니다. 또한 4-페닐 부티르산 또는 타우린 복합 우르소데옥시콜산과 같은 화학적 샤페론을 경구 투여하면 응급실 스트레스를 완화하고 인슐린 신호 전달을 개선하며 산부인과 쥐의 인슐린 감수성을 회복할 수 있습니다[141]. 간에서 발생하는 것과 마찬가지로 인슐린 분비에 대한 수요 증가는 만성 고혈당증 당뇨병 환자와 생쥐의 췌장 β세포에서 ER 스트레스를 유발하며, 이는 T2DM의 발달에 기여합니다[142,143]. ER 스트레스의 주요 전사 인자인 X-box-binding protein-1(XBP-1)의 췌장 β세포 특이적 녹아웃은 마우스에서 β세포 기능 장애로 인한 고혈당증 및 식이 유도 인슐린 저항성을 초래했습니다[139,144]. 따라서 ER 스트레스가 직접 인슐린 저항성을 유발한다고 결론짓는 것은 시기상조인 것으로 보이지만, ER 스트레스의 일부 측면은 지방 형성, 지질 방울 형성 및 지질 저장과 같은 포도당 및 지질 대사를 조절하는 것으로 보입니다.
- Other potential mechanisms for insulin resistance. Other hypotheses, such as endoplasmic reticulum (ER) stress, reactive oxygen species (ROS), and inflammation, have been proposed to explain the mechanism responsible for obesity-induced insulin resistance. PIP2, phosphatidylinositol-4,5-bisphosphate; PIP3, phosphatidylinositol-3,4,5-trisphosphate; PKB, protein kinase B; IKK, inhibitor of nuclear factor κ-B kinase; JNK, c-Jun N-terminal kinase; PKC, protein kinase C; LIPIN2, phosphatidic acid phosphatases; DAG, diacylglycerol; HFD, high-fat diet. Download Figure
- 사이토카인 생성의 변화와 염증 신호 전달 경로의 활성화를 특징으로 하는 전신 만성 염증 반응은 비만 관련 인슐린 저항성에 대한 역할을 결정하기 위해 활발히 연구되고 있습니다[145]. 종양괴사인자-α(TNFα)와 같은 염증 관련 사이토카인은 비만 및 당뇨병 인간과 동물의 지방세포에 의해 모집된 대식세포에 의해 배설되는 것으로 보고되었습니다[146,147]. 단핵구 화학유인 단백질 1(MCP1, CCL2라고도 알려진 케모카인 리간드)의 과발현으로 인해 대식세포가 지방 조직에 의해 모집될 때, 지방 조직에서 TNFα 발현이 증가하면서 간 인슐린 저항성이 관찰되지만 체중이나 비만도의 변화는 없습니다[148]. 또한 MCP1의 녹아웃은 HFD 유도 인슐린 저항성으로부터 보호되었습니다[148]. 일관되게 CCR2(MCP-1의 수용체)를 녹아웃하고 INCB3344(CCR2 길항제)를 사용한 치료는 인슐린 감수성을 증가시키고 지방 조직에 의한 대식세포 모집을 감소시켰습니다[149]. 인슐린 저항성에서 TNFα의 역할은 IRS1의 세린 307을 인산화하는 JNK1에 의해 매개되는 것으로 간주됩니다[150]. TNFα는 또한 핵인자 κ-B 키나아제(IKK)의 억제제를 활성화하고, 간 또는 골수성 세포 특이적 결실을 통해 간 인슐린 저항성으로부터 보호하고, 간 인슐린 신호 전달을 개선하며, 염증성 사이토카인 발현을 감소시키며[151], 이는 IKK가 인슐린 저항성에서 TNFα의 역할을 매개할 수 있음을 시사합니다. 이러한 연구는 인슐린 저항성에서 적어도 TNFα와 MCP1의 관여와 같은 염증의 역할을 뒷받침합니다.
- 그러나 NF-κB 필수 조절제(NEMO)의 간 특이적 녹아웃은 IKK 활성화, 공복 혈장 포도당 및 공복 인슐린 감소, 포도당 내성 개선, 간 염증 증가로 이어졌습니다[152]. 또한, NF-κB p65 서브유닛의 과발현은 식이 유발 인슐린 저항성을 어느 정도 보호하고 간 및 말초 인슐린 감수성을 개선했습니다[153]. 또한, TNFα 및 인터루킨-6과 같은 염증성 사이토카인의 혈장 수치는 T2DM의 인슐린 저항성 자손에서 변하지 않았으며, 인슐린 내성 노인 피험자는 인슐린 민감성 대조군과 비교하여 유사했습니다[154,155]. 이러한 연구는 만성 염증이 인슐린 저항성의 주요 원인 요인이 아니며 전신 포도당 대사를 방해하기에 충분하지 않다는 것을 보여줍니다. 따라서, 만성 염증은 간접적으로 인슐린 저항성을 악화시키는 것으로 보이며, 인슐린 저항성 및 T2DM의 주요 전략적 표적이 되어서는 안 된다. 인슐린 저항성과 포도당 대사에서 만성 염증의 역할을 더 잘 결정하기 위해서는 엄격하게 통제된 추가 연구가 필요합니다.
- T2DM 치료용 의약품
- 100년 전만 해도 당뇨병은 지속적인 체중 감소, 쇠약, 케톤산증, 그리고 결국 혼수상태와 사망을 동반하는 무서운 질병으로 여겨졌습니다. 항당뇨병제로 인슐린을 사용한 후 당뇨병 관련 사망률이 크게 감소했습니다. 그러나 주로 앉아서 생활하고 비만 유병률이 높아지면서 T2DM의 발병률이 증가했다[156]. T2DM을 개선하기 위해 생활습관 교정과 체중 감량이 권장되지만, 실현 가능성이 낮아 효과가 제한적입니다. 현재 T2DM에 처방되는 약물은 인슐린 분비를 자극하거나 인슐린 감수성을 증가시킨다(그림 6). 현재 사용 가능한 T2DM 치료용 의약품은 많은 문헌고찰에서 잘 설명되었으므로, 본 문헌고찰에서는 대표적인 T2DM 치료제를 간략하게 정리하였다.
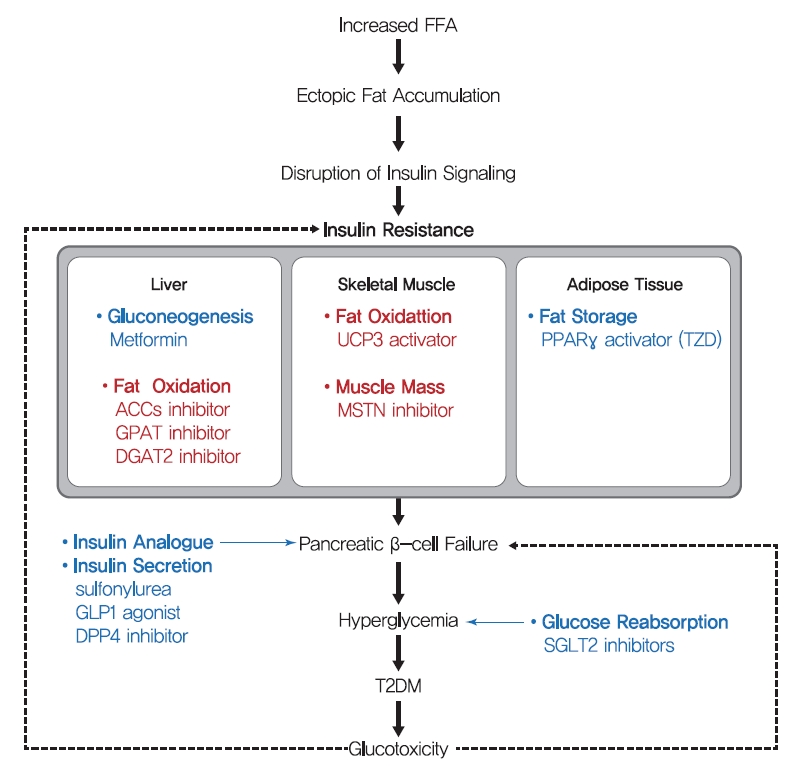
- 제2형 당뇨병(T2DM)의 개략적인 메커니즘과 인슐린 저항성에 대한 치료 전략. T2DM에 대한 현재(파란색) 치료와 인슐린 저항성에 대한 가능한 미래(빨간색) 치료의 주요 전략이 요약되어 있습니다. 설포닐우레아, 글루카곤 유사 펩타이드 1(GLP-1) 작용제, 디펩티딜 펩타이드-4(DPP-4) 억제제와 같은 많은 T2DM 약물은 β세포의 인슐린 분비 능력을 표적으로 합니다. 또한 티아졸리디네디온(TZD)과 메트포르민은 인슐린 감작 항당뇨병제로, 각각 간 내 지방 조직의 지방 저장 능력과 포도당 생성을 목표로 합니다. 본 연구에서 제시된 인슐린 저항성 치료의 주요 전략은 간과 골격근의 β 산화 증진과 근육 품질 증진을 목표로 하는 것이다. FFA, 유리 지방산; ACC, 아세틸-CoA 카르복실라아제; GPAT, 글리세롤-3-포스페이트 아실트랜스퍼라제; DGAT2, 디아실글리세롤 아실전이효소 2; UCP3, 분리 단백질 3; MSTN, 미오스타틴; PPARγ, 과산화소체 증식제-활성화 수용체-γ; SGLT2, 나트륨-포도당 공동수송체 2. Figure 다운로드
- β세포 기능 장애는 당뇨병 전단계에서 T2DM으로 전환되는 과정에서 중요한 특징이며[157], 많은 T2DM 약물은 β세포가 인슐린을 분비하는 능력을 표적으로 삼습니다. 설포닐우레아는 β세포에서 ATP에 민감한 K+ 채널의 조절 서브유닛인 설포닐우레아 수용체-1(SUR-1)을 차단하여 β세포가 포도당과 독립적으로 인슐린을 방출하도록 자극합니다[158]. 또한 글루카곤 유사 펩타이드-1(GLP-1)은 소장에서 L세포가 음식 섭취에 반응하여 생성하는 호르몬으로 췌장 β세포에 의한 인슐린 생성과 분비를 자극하고 췌장 α세포에 의한 글루카곤 분비를 억제합니다. GLP-1은 안정성이 낮기 때문에 반감기가 긴 GLP-1 작용제와 디펩티딜 펩타이드-4(DPP-4) 억제제를 사용하여 T2DM을 치료합니다. 이러한 약물은 β세포의 인슐린 분비를 자극하여 고혈당을 개선하고 포도당 독성으로 인한 위험을 줄이지만 질병 진행을 예방하지는 못합니다.
- 티아졸리디네디온(TZD)(예: 피오글리타존, 로시글리타존)은 골격근, 간, 지방 조직의 인슐린 감수성을 강화하고 간과 골격근에서 지방세포로의 지방 재분배를 촉진합니다[159]. 인슐린 감수성에 대한 TZD의 영향은 포도당 및 지질 대사 및 에너지 균형에 관여하는 여러 유전자의 전사를 조절하고 지방 산화, 지방 세포 증식, 지방 형성, 지방 재분포를 증가시키고 혈장 유리 지방산 및 전염증성 사이토카인 수치를 감소시키는 핵 수용체인 PPARγ의 활성화에 의해 매개됩니다[159]]. 당뇨병에 가장 흔하게 처방되는 약물인 메트포르민은 HGP를 감소시켜 공복 중 말초 조직의 인슐린 감수성을 증가시킬 수 있으며, HGP는 미토콘드리아 활성의 촉진 또는 AMPK 활성화를 통한 글루카곤 신호전달의 억제에 의해 매개되는 것으로 간주됩니다[160,161].
- In addition to drugs that increase insulin secretion and sensitivity, some T2DM drugs use other strategies. Alpha-glucosidase inhibitors (AGIs) inhibit gastrointestinal hydrolyzing enzymes, which delay the absorption of carbohydrates in small intestine and decrease postprandial blood glucose and insulin levels [162]. On the other hand, sodium-glucose cotransporter 2 (SGLT2) inhibitors lower blood glucose levels by blocking renal glucose reabsorption, which reduces glucotoxicity, improves β-cell function, and increases insulin sensitivity [163].
- 위에서 지적한 바와 같이 당뇨병 치료를 위해 많은 약물이 처방되고 있으며, 당뇨병 치료제 개발과 관련된 막대한 의료비는 계속해서 제약 회사에 부담을 주고 있습니다. 그럼에도 불구하고 최근 몇 년 동안 당뇨병의 유병률과 사망률이 급격히 증가했습니다. T2DM의 주요 원인인 인슐린 분비 장애 및 인슐린 저항성을 고려할 때, 현재 제약 전략의 명백한 비효율성은 인슐린 분비를 자극하여 고혈당증과 같은 증상을 완화하는 데 지나치게 집중했기 때문일 수 있습니다. 또한 인슐린 저항성을 표적으로 하는 약물은 2개에 불과한 반면, 인슐린 분비를 표적으로 하는 약물은 많다[164]. 증거에 따르면 인슐린 저항성은 T2DM 발병의 선행 및 중심이며 주로 조직 내 이소성 지방 축적에 의해 주도됩니다. 또한 인슐린 저항성은 비만과 밀접한 관련이 있는 대사 증후군 발병의 일반적인 병원성 요소입니다. 따라서 우리는 인슐린 저항성을 표적으로 하는 다양한 치료 전략을 채택할 것을 제안하며, 이 접근법이 당뇨병과 대사 증후군을 예방하고 치료하는 보다 효과적인 수단을 제공한다고 믿습니다.
- 인슐린 저항성에 대한 치료 전략
- 위에서 언급한 바와 같이, 간세포에 지질이 축적되면 간 DAG에 의한 PKCε의 활성화를 통해 간 인슐린 저항성을 유도합니다. 간 기능은 포도당과 지질 대사 조절의 핵심이기 때문에 간 인슐린 저항성은 전신 인슐린 저항성으로 이어집니다. 따라서, de novo lipogenesis 및 re-esterification를 통한 지질 합성의 억제는 간에서 지질 축적을 감소시키는 효과적인 수단을 구성하는 것으로 생각된다. 이 의견은 동물 실험 결과에 의해 잘 뒷받침됩니다. ACC1과 ACC2는 각각 새로운 지방 형성 및 지질 산화의 중요한 조절자입니다. ASO를 사용한 ACC1 및 ACC2 억제는 간 DAG 함량과 PKCε 원형질막으로의 전위를 감소시키고 지질 유도 간 인슐린 저항성으로부터 마우스를 보호했습니다[165]. 또한 아데노바이러스에 의한 미토콘드리아 글리세롤-3-포스페이트 아실전이효소(mGPAT, 지방형성 속도 제한 효소)의 과발현은 인슐린 저항성을 유발하고 간세포의 DAG 수치를 증가시켰습니다[166]. 같은 맥락에서, mGPAT 녹아웃 마우스는 간 DAG 수치를 감소시키고 HFD 유도 간 인슐린 저항성으로부터 보호되었으며[167], 포스파티드산 인산분해효소(PAP, Lipins라고도 함)의 shRNA 녹다운은 유사하게 간 DAG 수치를 감소시키고 인슐린 감수성을 개선한 반면, 아데노바이러스를 사용한 리핀-2의 과발현은 포도당 내성과 인슐린 감수성을 교란시켰다[168,169]. 또한, 디아실글리세롤 아실전이효소 2(DGAT2, 디아실글리세롤에서 트리글리세라이드 합성의 마지막 단계를 촉매하는 효소)의 억제는 간 DAG 함량을 감소시키고, 식이 유도 간 지방증 및 인슐린 저항성을 역전시키며, 원형질막의 PKCε 수치를 감소시켰습니다[170]. 이러한 연구는 간신지방형성을 억제하여 간 DAG 및 아세틸-CoA 함량을 감소시키는 치료법이 T2DM에서 간 인 슐린 저항성을 효과적으로 치료할 수 있다는 주장을 강력하게 뒷받침합니다. 이 전략에서는 ACC1, ACC2 및 DGAT2를 표적으로 하는 약제가 T2DM 및 비알코올성 지방간염 치료를 위한 임상 2상을 진행 중입니다(표 1) [171].
- 현재 간에서 지방 합성을 억제하고 골격근에서 지방 산화 및 근육량을 자극하는 전략에 따라 인슐린 저항성을 표적으로 하는 약물을 개발 중입니다.
데이터베이스 Cortellis Drug Discovery Intelligence(클래리베이트 애널리틱스)의 데이터[171].
ACC, 아세틸-CoA 카르복실라아제; DGAT2, 디아실글리세롤 아실전이효소 2; PPAR, 과산화소체 증식제-활성화 수용체; MSTN, 미오스타틴.
- 간은 포도당 대사에 질적으로 중요하지만, 골격근은 체중의 35%-45%, 인슐린 자극 후 총 포도당 처리량의 최대 70%-80%를 차지하기 때문에 골격근은 정량적으로 필수적입니다. 그러므로, 지방 산화 및/또는 골격근량을 증가시키는 것은 intrayocellular 지질 축적을 감소시키고, 결국에는, 인슐린 감수성을 개량하기 위한 잠재적인 전략이라고 여겨진다. 골격근에서 HFD 유도 인슐린 저항성으로부터 보호되는 분리 단백질 3(UCP-3, 미토콘드리아 양성자 구배를 소멸시키는 내부 미토콘드리아 막 수송체)의 골격근 특이적 과발현, 에너지 소비 증가, PKCθ 활성 및 막 관련 DAG 수준 감소[172]. 근육의 지방 산화가 인슐린 저항성에 미치는 유익한 효과는 ACC-2(미토콘드리아 지방 산화의 핵심 조절자)를 쓰러뜨린 쥐에서도 관찰되었습니다[173]. 또한, 자가포식 관련 유전자-7(ATG-7)의 골격근 특이적 결실은 식이요법으로 인한 비만과 인슐린 저항성으로부터 마우스를 보호했으며, 지방 산화 증가와 지방 세포 갈변을 동반했습니다[174]. 또한, PPARδ의 골격근 특이적 과발현은 포도당 내성을 개선하고 미토콘드리아 산화 대사가 에너지 생산에 기여하는 느린 경련 근섬유(미토콘드리아가 풍부한 섬유 유형)의 수를 증가시켰습니다[175]. 또한, 마이오스타틴(MSTN, Gdf-8이라고도 함) 절제에 의해 유도된 빠른 경련 근육 섬유의 수가 증가하거나 구조적으로 활성화된 형태의 Akt1의 골격근 특이적 발현은 식이요법 유발 비만을 예방하고, 말초 인슐린 감수성을 향상시켰으며, 식이 유도 인슐린 저항성을 극적으로 개선했으며[176], 이는 아마도 근육량 증가로 인한 포도당 이용 증가 및 지방 산화에 기인했을 것입니다. 근육량 증가와 포도당 대사 개선의 유익한 효과에도 불구하고 개발 중인 당뇨병 치료제는 근육을 표적으로 합니다. MSTN에 대한 단클론 항체는 대사 장애 및 심혈관 질환 치료를 위한 임상 1상 시험 중입니다(표 1). 근육의 질을 높이기 위한 발달 노력과 관련하여, 간경변증 치료에 사용되어 왔으며 PPAR을 표적으로 하는 베자피브레이트는 T2DM에 대한 2상 임상시험의 대상이며, PPAR 범작용제인 치글리타자르 나트륨은 최근 중국에서 T2DM 치료제로 승인되었습니다(표 1) [171].
인슐린 저항성에 대한 치료 전략

간에서 지방 합성 억제
다운로드 표지방 산화와 근육량의 자극
- 세계보건기구(WHO)에 따르면 당뇨병 환자 수는 1980년에서 2019년 사이에 1억 800만 명에서 4억 6,300만 명으로 거의 4배 증가했으며, 이러한 증가는 비만 유병률의 꾸준한 증가를 동반했습니다. 또한 당뇨병으로 인한 조기 사망률은 2000년에서 2016년 사이에 5% 증가했습니다. 대부분의 국가에서 고혈당증의 유병률은 7%에서 14% 사이이다[156]. 또한 전 세계 당뇨병 유병률은 2045년까지 7억 명으로 증가할 것으로 예상되며, 대사 질환의 유병률이 급격히 증가함에 따라 현재 글로벌 보건 비상사태로 간주되고 있습니다[164].
- 대사 질환의 주요 원인인 인슐린 저항성은 당뇨병을 포함한 대사 질환의 치료 대상으로 간주되어야 합니다. 그러나 일반적으로 받아들여지는 이론은 인슐린 저항성의 메커니즘을 설명하지 못합니다. 그럼에도 불구하고 이소성 지질 축적이 ER 스트레스 및 염증성 사이토카인의 혈장 농도와 같은 다른 변수보다 당뇨병 생리학과 더 밀접한 관련이 있다는 증거가 증가하고 있습니다[110,111,126]. 수많은 동물 및 역학 연구에 따르면 이소성 지방 축적 또는 지방 산화 감소로 인한 원형막 분획의 DAG 축적은 간 조직 및 골격근에서 nPKC의 활성화를 통한 인슐린 저항성 발달의 중요한 요소입니다. 이러한 인슐린 저항성의 병태생리학적 기전을 바탕으로 간에서 지질 합성을 억제하고 골격근에서 지방 산화를 자극함으로써 이소성 지질 축적을 줄이고 잠재적으로 인슐린 감수성을 개선하여 궁극적으로 T2DM의 발병을 예방하거나 지연시킬 수 있습니다./ 출처 당뇨병학회
결론